신규 약물 설계를 위한 인공지능 기술 동향
Technical Trends in Artificial Intelligence for De Novo Drug Design
- 저자
-
한영웅의료정보연구실 hanhero@etri.re.kr 정호열의료정보연구실 hoyoul.jung@etri.re.kr 박수준디지털바이오의료연구본부 psj@etri.re.kr
- 권호
- 38권 3호 (통권 202)
- 논문구분
- 지능형 미래사회 구현을 위한 디지털 융합 기술
- 페이지
- 38-46
- 발행일자
- 2023.06.01
- DOI
- 10.22648/ETRI.2023.J.380305
 본 저작물은 공공누리 제4유형: 출처표시 + 상업적이용금지 + 변경금지 조건에 따라 이용할 수 있습니다.
본 저작물은 공공누리 제4유형: 출처표시 + 상업적이용금지 + 변경금지 조건에 따라 이용할 수 있습니다.- 초록
- The value of living a long and healthy life without suffering has increased owing to aging populations, transition to welfare societies, and global interest in health deriving from the novel coronavirus disease pandemic. New drug development has gained attention as both a tool to improve the quality of life and high-value market, with blockbuster drugs potentially generating over 10 billion dollars in annual revenue. However, for newly discovered substances to be used as drugs, various properties must be verified over a long period in a time-consuming and costly process. Recently, the development of artificial intelligence technologies, such as deep and reinforcement learning, has led to significant changes in drug development by enabling the effective identification of drug candidates that satisfy desired properties. We explore and discuss trends in artificial intelligence for de novo drug design.
Share
Ⅰ. 서론
코로나19 바이러스로 인해 빠른 신약 개발의 중요성을 체감하면서 국내외 제약기업들의 기술력 확보가 강조되고 있다. 신약의 경우 물질특허가 가능하고, 제품의 생명력이 길고, 신흥 시장에서의 잠재력이 높으며 소재에 대한 의존도가 높지 않기 때문에 한국과 같이 자원이 부족한 국가에서 매력적인 시장이다. 2020년 기준으로 의약품 허가 및 신고된 품목 중 신약은 40개인데, 이중 국내 개발 신약은 개량 신약 6종에 불과한 것으로 나타나 국내외 시장 경쟁력을 갖추기 위해서는 신약 개발 기술에 대한 체질 개선이 필요하다.
신규 약물 개발 프로세스는 후보물질 발굴, 전임상 및 임상 시험의 단계로 구성되는데, 후보물질 발굴 단계에서 약물로 활용될 수 있는 신규 화합물을 발굴한다. 이론적으로 1030개 이상의 신규 화합물을 합성할 수 있다고 알려져 있기 때문에 원하는 특성을 갖는 화합물을 발견하는 것은 매우 어려운 일이다. 이로 인해 신약 발굴에서부터 출시까지 평균 10년 이상이 소요되며, 전체 신약 개발 비용 중 1/3 이상이 후보물질 발굴 단계에서 소요된다. 화합물 라이브러리로부터 1만 개의 물질을 스크리닝 하게되면 임상 시험까지 들어가는 경우는 평균 10개 정도이고, 임상 시험에 진입한 물질도 시판 승인까지 성공한 케이스는 10% 미만으로 알려져 있다.
최근 딥러닝(Deep Learning)과 같은 인공지능(AI: Artificial Intelligence) 기술의 발달과 알려진 화합물 분자 구조에 대한 데이터의 지속적인 증가로 신약 개발 분야에서도 인공지능 기술이 빠르게 도입되고 있다. 특히 인공지능을 이용한 후보물질 발굴은 전통적인 신약 개발 프로세스에서와 같이 한정된 화합물 라이브러리에서 후보물질을 스크리닝하는 것이 아니라 합성 가능한 모든 화합물을 고려하여 후보물질을 새롭게 생성하는 신규 약물 설계(De novo drug design) 기술의 등장으로 패러다임의 변화를 맞이하고 있다. 인공지능 기반의 신규 약물 설계를 통해 후보물질을 효과적으로 발굴할 수 있어서 오랫동안 큰 변화가 없었던 신약 발굴 소요 기간 및 비용을 획기적으로 감축할 수 있을 것으로 기대를 모으고 있다. 이와 같은 기술의 변화에 따라 글로벌 인공지능 신약 시장은 2025년까지 연평균 50%의 높은 성장률을 보이며, 약 30억 달러(약 4조 원)의 규모로 성장할 것으로 예측되고 있다[1]. 이에 본고에서는 신약 개발 프로세스의 큰 변화를 주도하고 있는 신규 약물 설계를 위한 인공지능 기술에 대한 연구 동 향을 살펴보고자 한다.
Ⅱ. 현재의 인공지능 기반 신약 설계
1. 신약 개발 프로세스
신약 개발 프로세스는 그림 1(a)와 같이 크게 3단계로 구성된다. 첫 번째는 알려지지 않은 신약 후보물질을 찾는 후보물질 발굴(Discovery and Development) 단계이다. 후보물질 발굴 단계는 그림 1(b)와 같이 먼저 Target Identification을 통해 질환 연관 타겟 단백질을 선정하고, Compound Design을 통해 인공지능을 기반으로 원하는 특성을 갖는 후보물질을 설계한다. 전통적인 신약 설계 프로세스에서는 후보물질 탐색을 위해 라이브러리 기반의 화합물 스크리닝을 수행하는데, 인공지능 기반 신약 설계는 화합물 스크리닝이 신약 설계로 대체된다. 신규 화합물이 설계되면 선도물질 도출(Hit-to-Lead) 및 실험적 검증(In vitro/In vivo Experiments)을 통해 QSAR(Quantitative Structure-Activity Relationship), 생리활성, 독성 등을 분석 및 검증하여 최종 후보물질을 도출한다. 이와 같이 신약 설계는 전체 신약 개발 프로세스의 출발점으로 이후 단계들에서 검증될 신약 후보물질을 생성/발굴하는 단계이므로 효과적으로 승인 가능성이 큰 후보물질을 발굴하는 것이 전체 신약 개발 프로세스의 소요 기간 및 비용을 단축하는 데 중요한 역할을 한다.
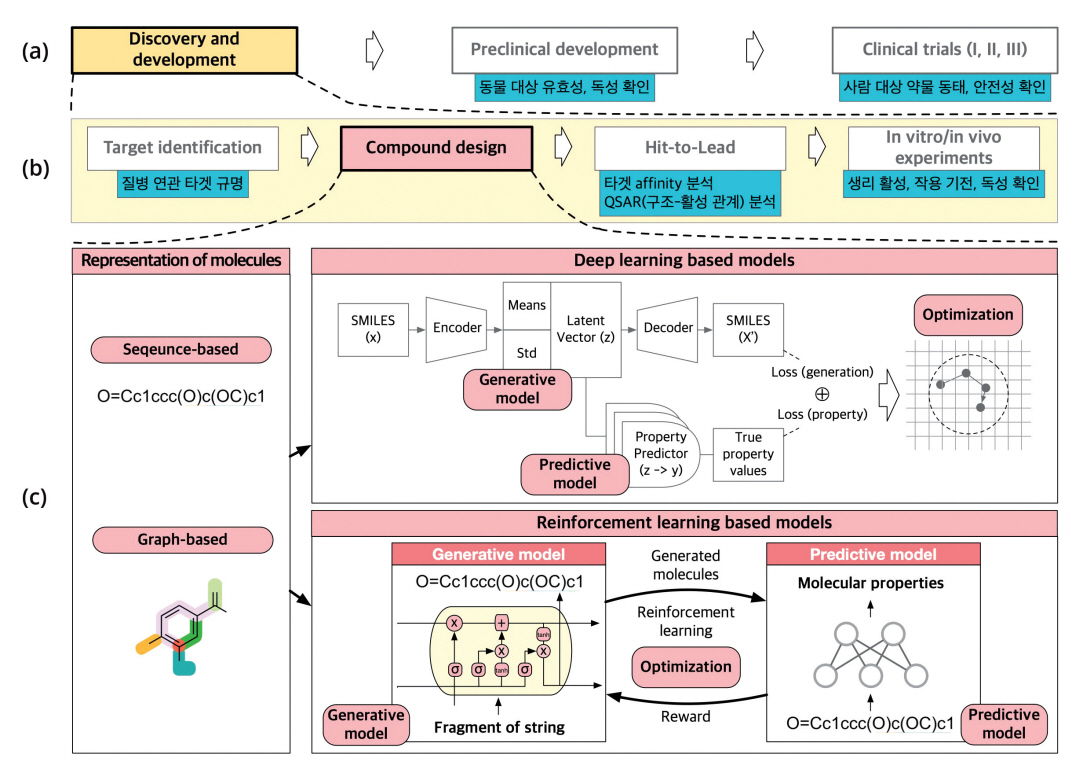
현재의 인공지능 기반 신약 설계는 그림 1(c) [2]와 같이 화합물의 분자 구조를 인공지능에 입력할 수 있는 형태로 나타내는 표현 모델링(Representation of Molecules), 기존 화합물의 특징을 학습하여 새로운 화합물을 만들 수 있는 생성 모델(Generative Model), 화합물의 물리화학적/약리학적 특성 및 생리학적 활성값을 정량화할 수 있는 예측 모델(Predictive Model), 생성 모델과 예측 모델을 동시에 최적화하여 원하는 특성을 갖는 최적의 신규 약물 후보군을 도출할 수 있는 최적화 기술(Optimization)로 구성된다. 이 장에서는 각 세부기술에 대한 최신 동향을 소개하고 자 한다.
2. 화합물 구조 표현 모델링 기술
인공지능 모델이 화합물의 분자 구조를 학습하여 새로운 화합물을 출력하기 위해서는 인공지능이 이해할 수 있는 형태로 화합물의 분자 구조를 변환해야 하는데, 이를 표현 모델링이라고 한다. 표현 모델링은 Sequence 기반의 방법과 Graph 기반의 방법이 가장 많이 활용되고 있다(그림 1(c)). Sequence 기반의 표현 모델링 방법은 화합물을 구성하는 분자를 특정 순서에 따라서 String 형태로 기록하는 표기법으로 SMILES(Simplified Molecular Input Line Entry System)가 대표적이다[3]. String 형태이므로 다양한 기계학습 알고리즘에 활용하기 편하다는 장점이 있으나 유사한 분자 구조를 상당히 다른 결과물로 표현할 수 있다는 한계가 있다[4]. Graph 기반의 표현 모델링 방법은 원자와 공유결합 정보가 각각 Node와 Edge로 구성된 Vector 형태로 화합물의 구조를 표현하는 방법이다. 이는 Sequence 기반 표현 모델링 방법보다 직관적이고 실제 화합물의 분자 구조에 가까운 표현이 가능하나, Graph 형태의 데이터를 입력받을 수 있는 기계학습 방법에만 입력이 가능하다는 한계가 있다. 대표적으로 분자를 정해진 Sub-structure의 집합으로 생각해서 처리하는 Junction tree 방식이 있다[5].
3. 신규 약물 후보군 생성 기술
신약이 될 수 있는 새로운 화합물을 발굴하기 위해서는 알려진 약물에 대한 학습 데이터셋을 구축하고, 이를 인공지능 기반 생성 모델(Generative Model)에 입력하여 약물로 활용되는 화합물들의 분자 구조 특성을 학습한 후, 학습된 모델을 이용해 새로운 약물 후보군을 출력한다. 최근 연구들은 생성 모델을 구축하기 위해 주로 VAE(Variational Auto-Encoder), RNN(Recurrent Neural Network), GAN(Generative Adversarial Network) 방법을 이용한다[6]. VAE는 입력 데이터를 압축 및 복원할 수 있는 Latent Vector를 만드는 Auto-Encoder가 변형된 형태로 입력 데이터를 표현할 수 있는 분포를 학습하여 이 분포로부터 학습 데이터와 유사한 데이터를 생성해낼 수 있는 인공지능 기술이다. RNN은 시간이나 순서에 따라 기록된 데이터를 분석하는 데 뛰어난 성능을 발휘하는 딥러닝 방법으로 SMILES와 같이 String 형태로 표현된 분자 구조의 특징을 학습하여 새로운 화합물을 생성해낼 수 있다. GAN은 새로운 화합물을 만들어 내는 Generator와 만들어진 화합물이 실제로 존재하는 화합물인지 만들어진 화합물인지 판별하는 Discriminator가 서로 대립하며 학습함으로써 성능을 점차 개선해 나가는 방법이다.
4. 특성 정량화 예측 기술
생성 모델에서 만들어진 화합물이 신약이 되기 위해서는 약물이 가져야 하는 여러 가지 특성을 만족해야 하므로 화합물의 특성을 정량화할 수 있는 기술이 필요하다. 약물로 사용되기 위해서 갖춰야 할 특성을 Drug-likeness라고 하는데, Lipinski’s rule of five가 가장 많이 활용된다[7].
● 수소결합 Donor의 개수는 5개 이하
● 수소결합 Acceptor의 개수는 10개 이하
● 분자량은 500Daltons 이하
● LogP(분배계수)는 4 이하
Drug-likeness 외에도 신약으로 활용되기 위해서는 Novelty(기존에 없던 화합물이어야 함), Synthesizability(합성 가능해야 함), Potency at the target(타겟에 대한 활성이 존재해야 함) 등 여러 가지 특성을 만족해야 하는데, 화합물의 구조를 기반으로 직접적으로 계산될 수 없는 특성들은 인공지능 기반의 특성 정량화 예측 기술을 통해 특성값을 예측해야 한다.
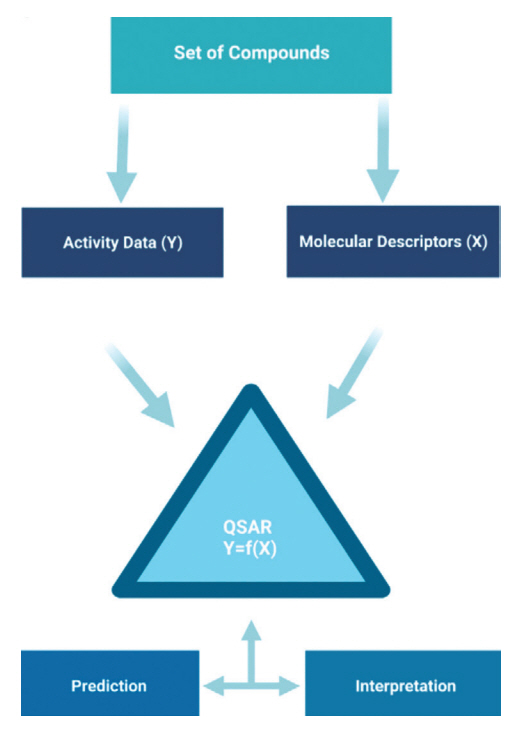
인공지능 기반의 특성 정량화 예측 기술은 QSAR (Quantitative Structure-Activity Relationship)과 유사한 방법으로, 알려진 분자 구조와 특성값의 연관관계를 인공지능 모델을 이용해 학습하여 새로운 화합물의 분자 구조를 기반으로 특성값을 예측할 수 있는 모델을 구축하고, 이를 이용해 생성 모델에서 생성한 새로운 화합물 분자 구조에 대한 특성값을 예측하는 기능을 수행한다(그림 2) [8].
5. 최적화 기술
최적화 기술은 생성 모델과 예측 모델의 손실값들을 통합하여 Gradient 기반으로 최적화하는 방법과 예측 모델의 예측값을 Reward로 사용하여 강화학습을 기반으로 최적화하는 방법이 사용된다(그림 1(c)). AlphaGo를 비롯해 게임 등 다양한 분야에서 강화학습이 우수한 성능을 보이고 있는데, 이를 반영하여 신약 설계 분야에서도 강화학습을 활용한 연구가 점점 늘어나는 추세이다. 강화학습은 목표 지점까지의 최적 경로를 찾는데 우수한 성능을 나타내기 때문에 순차적으로 원자를 추가하여 화합물의 분자 구조를 만들어나가는 방식의 생성 모델들이 강화학습 기반의 최적화 기술과 조합되어 활용되는 양상을 나타낸다.
Ⅲ. 미래의 인공지능 기반 신약 설계
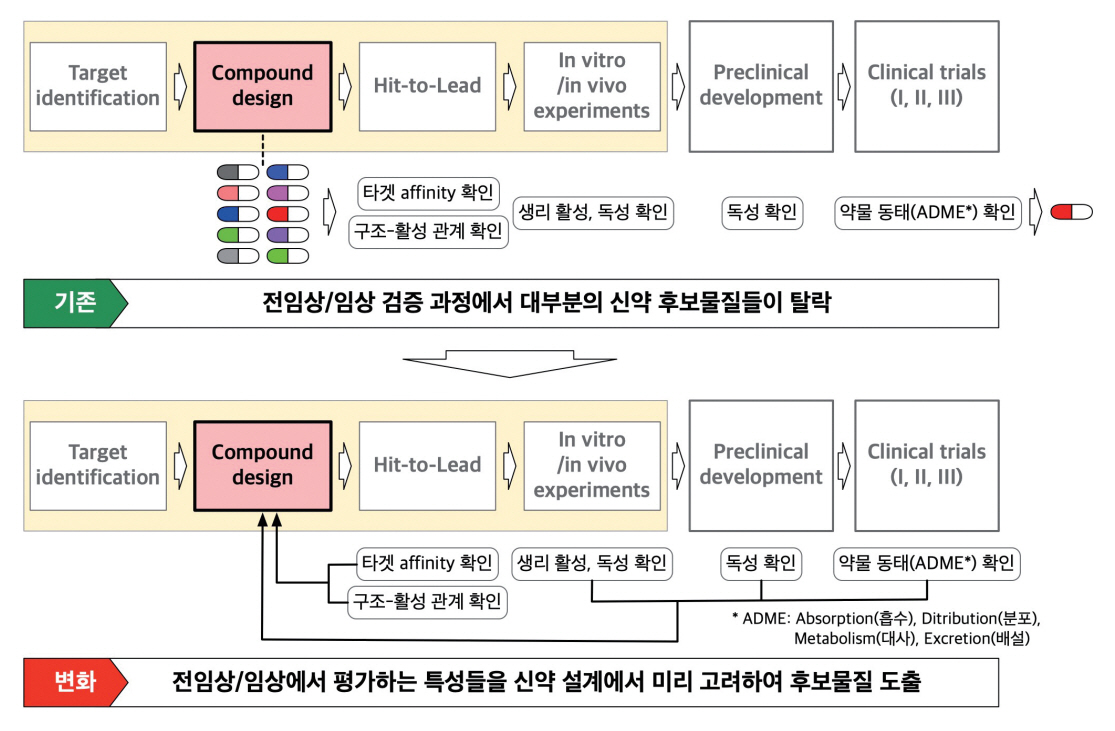
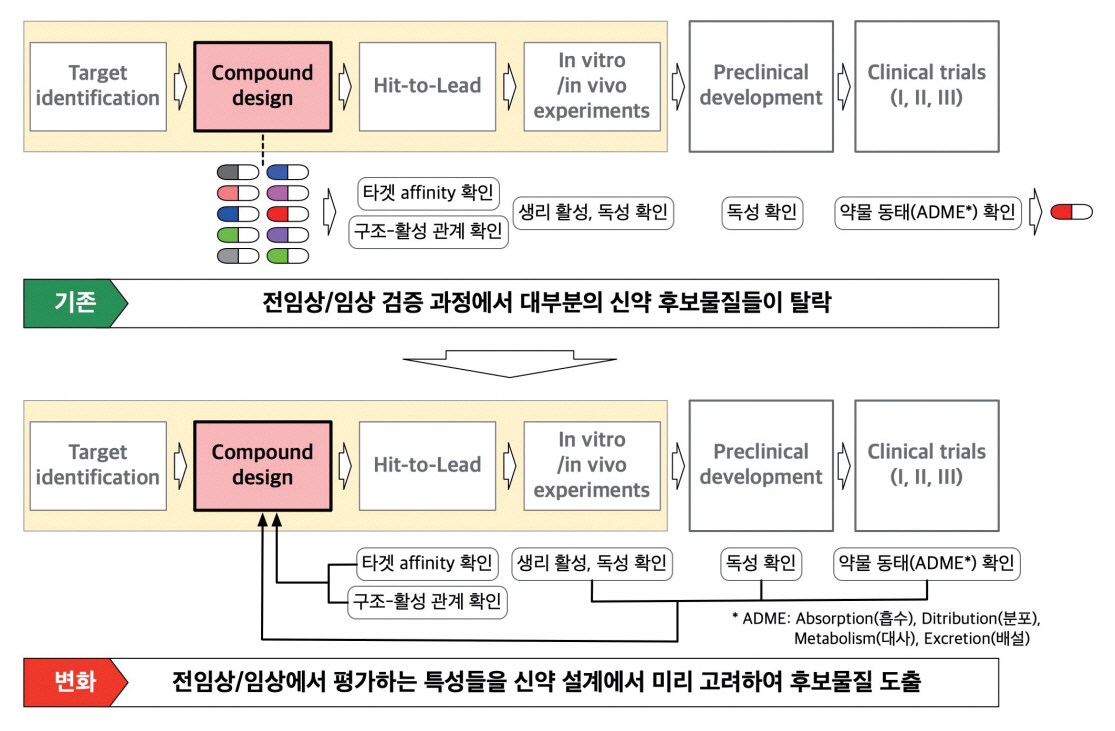
전통적인 신약 개발 프로세스는 먼저 후보물질을 발굴하고, 이후의 전임상 및 임상 단계에서 발굴된 후보물질의 특성을 검증한다. 특성을 검증하는 과정에서 타겟과의 결합 친화도가 떨어지거나, 합성이 어렵거나, 독성이 있거나, 안정성에 문제가 있다고 판단되면 신약으로 승인받을 수 없기 때문에 많은 후보물질이 전임상 및 임상 단계에서 탈락된다. 최근에는 인공지능 기술이 도입되면서 이와 같은 신약 개발 프로세스에 큰 변화가 나타나고 있다.
초기의 인공지능 기반의 신약 설계는 전통적인 화합물 라이브러리 기반 스크리닝을 대체하는 역할을 수행할 뿐 전체 신약 개발 프로세스에 변화를 가져오지는 못했다. 하지만 최근 연구들에서는 타겟 결합 특성이나 독성과 같은 신약 설계 이후의 단계들에서 평가되던 특성들을 신약 설계 단계부터 고려함으로써 후보물질들의 탈락 가능성을 현저히 낮추고 신약 개발의 효용성을 제고하려는 노력이 나타나고 있다. 이 장에서 인공지능 기술의 도입으로 인한 신약 설계 단계의 변화에 대한 움직임을 살펴보고자 한다.
1. 구체적인 타겟에 대한 신약 설계
본고의 Ⅱ장에서 설명한 바와 같이 신약 설계 단계에서는 Target Identification을 통해 질환 연관 타겟 단백질을 선정하고, Compound Design을 통해 원하는 특성을 갖는 후보물질을 설계한다. 새롭게 설계되는 후보물질은 타겟과 결합하여 기능을 나타내어야 하므로 타겟과의 결합 친화도는 신약 설계 단계에서 반드시 고려되어야 하는 특성이다.
초기의 인공지능 기반 신약 설계 연구는 기존 약물과 유사한 특성을 갖는 새로운 화합물의 생성에 초점을 맞췄기 때문에 구체적인 타겟을 고려하지 않거나 타겟을 고려할 때도 타겟과 후보물질 간의 직접적인 결합보다는 활성 억제 효과와 같은 결합에 관한 간접적인 정보를 고려했다. 2018년에 발표된 ReLeaSE [9] 연구는 강화학습 기술을 이용해 생성 모델과 물리화학적 특성 예측 모델을 최적화함으로써 인공지능 기반 신약 설계에 대한 기반 기술을 정립하였는데, 해당 연구에서도 타겟인 JAK2(Janus Protein Kinase 2)에 대한 억제물질의 발굴을 위해서 JAK2의 기능을 억제하는 물질의 효능을 측정한 값인 IC50(Half maximal inhibitory concentration, 반수 최대 억제 농도)을 활용하여 타겟 결합 특성을 고려하였다. 2019년에 인공지능 신약 개발 기업인 Insilico medicine 사에서 발표한 GENTRL [10] 연구는 DDR1(Discoidin Domain Receptor 1)의 억제 후보 물질을 발굴하였는데, 기계학습 기술인 SOM(Self Organizing Map)을 이용해 알려진 DDR1 억제물질들과의 유사도를 측정함으로써 간접적으로 타겟과의 결합을 고려하였다.
최근에는 화합물의 분자 구조를 이용해 타겟과의 결합을 예측할 수 있는 인공지능 기술들이 발표되고 있고, 예측된 타겟 결합 친화도를 신약 설계에 활용하는 연구도 수행되고 있으므로 가까운 미래에는 신약 설계 단계에서 타겟과의 결합을 고려한 후보물질의 발굴이 가능할 것으로 예상된다. 2021년에 발표된 V-Dock [11] 연구는 화합물 구조에 대한 SMILES를 입력으로 받아 타겟과의 결합을 예측할 수 있는 인공지능 기술을 발표했고, 같은 해에 신약 설계 프레임워크인 REINVENT [12]를 개발한 연구팀은 구조 모델링 소프트웨어를 이용해 예측한 화합물의 3D 구조를 활용해 타겟 결합을 예측할 수 있는 DockStream [13]을 발표했다. AlphaGo를 개발한 DeepMind는 딥러닝을 기반으로 단백질의 구조를 예측하는 state-of-the-art 기술인 AlphaFold [14]를 발표했는데, 이를 이용하여 2022년에는 단백질-단백질 상호작용을 예측하는 연구가 발표되었다[15].
2. 전임상/임상을 고려한 신약 설계
신약 설계 단계에서 발굴한 신약 후보물질 중 대다수가 In vitro/In vivo 실험 및 전임상 단계에서 탈락된다. 후보물질들에 대한 높은 탈락률은 전체 신약 개발 프로세스의 효율성을 감소시키므로 효과적인 신약 개발을 위해서는 신약 설계 단계부터 이후 단계에서 검증될 특성들을 미리 고려하는 것이 필요하다.
초기의 인공지능 기반 신약 설계 연구는 Druglikeness를 만족하는 신규 화합물을 생성하는 데 초점을 맞췄기 때문에 독성(Toxicity)이나 약물 동태(ADME: Absorption, Distribution, Metabolism, Excretion)와 같이 전임상과 임상 단계에서 필수적으로 검증되는 특성들에 대해서는 고려하지 못했으나 최근 연구들에서 변화가 나타나고 있다. 2020년에 발표된 CogMol [16] 연구는 VAE를 기반으로 코로나 19 바이러스 단백질의 기능을 억제하는 물질을 찾는 프레임워크를 개발하였는데, 타겟 결합 특성과 Drug-likeness 특성을 고려한 인공지능 신약 설계 모델을 구축하였고, 이를 통해 생성된 후보물질들을 다시 선별하는 과정에서 딥러닝을 이용해 독성을 예측한 결과를 활용하였다. 2021년에 발표된 PaccMann [17] 연구에서는 신규 항암 물질을 찾기 위해서 VAE와 강화학습 방법을 이용해 IC50 값을 최적화함으로써 타겟 결합 특성을 고려할 수 있는 신약 설계 방법을 제안했는데, case study를 통해 타겟 결합 특성뿐만 아니라 독성도 함께 고려하여 후보물질을 발굴하는 연구를 시도하였다.
최근에는 약물 동태나 독성에 대해서 기계학습이 가능할 정도의 데이터를 모을 수 있는 환경이 갖춰졌기 때문에, 이를 이용한 예측 연구가 진행되고 있으므로 가까운 미래에는 전임상/임상에서 검증되는 약물의 특성들을 신약 설계 단계부터 고려하여 후보 물질을 발굴할 수 있을 것으로 예상된다(그림 3).
약물 동태 중 흡수(Absorption)의 경우 인간 장내 흡수(HIA: Human Intestinal Absorption)와 막 투과성(Membrane Permeability)을 통해 확인할 수 있는데, 해당 수치들은 인간 대장암(Caco-2) 세포주 투과성 분석 및 인공지질막 투과성 분석(PAMPA: Parallel Artifi cial Membrane Permeability Assay) 방법으로 측정 가능하므로[18] 최근에 이를 예측하는 연구들이 발표되고 있다. Lanevskij [19] 연구에서는 문헌 정보에서 수집된 Caco-2 세포의 투과성 데이터를 기반으로 투과성 예측을 위한 비선형 회귀모델을 개발했고, Sun [20] 연구에서는 인공지질막 투과성 분석 데이터를 활용하여 SVM(Support Vector Machine) 기반의 투과성 예측 모델을 개발하였다.
약물 독성은 대표적으로 간 독성(약물에 의한 간 손상), 심장 독성(심장박동 리듬에 관여하는 유전자의 억제로 인해 발생) 및 세포 독성이 있는데, Tox21 [21] 등의 데이터를 활용해 이와 같은 독성을 예측하는 연구들이 발표되고 있다. Kim [22] 연구에서는 베이지안 가중 지문 기술을 통해 간독성을 예측할 수 있는 모델을 개발하였고, Ogura [23] 연구에서는 유전자 알고리즘을 이용해 독성 예측에 활용될 입력 특징을 선별하고, SVM 모델을 이용해 심장 독성을 예측하였다. Cai [24] 연구에서는 Multi-tasking DNN(Deep Neural Network)을 이용해 심장 독성을 예측하였다.
Ⅳ. 결론
본고에서는 신규 약물 설계를 위한 인공지능 기술들의 현황과 인공지능 기술의 도입으로 인해 나타나고 있는 신약 설계 단계의 영역 및 역할에 대한 변화의 움직임에 대해서 살펴보았다. 신약 개발 프로세스는 평균 10년 이상의 기간이 소요되지만, 전임상 및 임상 시험을 통해 검증되고 선별되어 신약으로 승인되는 비율은 1% 미만으로 대부분의 후보 물질이 안전성(Safety) 및 효능(Efficacy) 측면에서 문제가 발견되어 탈락된다. 이와 관련된 특성인 독성이나 약물 동태 등을 신약 설계 단계부터 고려할 수 있다면, 후보물질들의 낮은 탈락률과 신약 개발 기간의 단축을 기대할 수 있으므로 인공지능 기술을 기반으로 한 신약 설계의 고도화에 대한 관심이 매우 높은 상황이다. 2018년 발표된 ReLeaSE [9] 연구를 시발점으로 생성 모델, 특성 예측 모델, 강화학습 기반 최적화 기술로 구성된 신약 설계 프레임워크가 정립되었고, 이를 기반으로 최근 연구들에서는 타겟 결합 특성이나, 전임상/임상에서 평가되는 약물 특성들을 신약 설계단계에서 미리 고려함으로써 승인 가능성 높은 신약 후보물질을 발굴하려는 연구들이 발표되고 있다. 인공지능 기술의 발전을 통해 가까운 미래에는 적중률 높은 신약 후보물질을 효과적으로 발굴할 수 있게 됨에 따라 연구 개발 비용을 획기적으로 절감하여 중소업체들의 진입장벽을 낮춤으로써 대형 제약회사 중심의 기존 관련 산업 생태계를 탈피하고, 이에 따라 자연스러운 시장 경쟁을 유도함으로써 의료 비용이 감소될 수 있을 것으로 예상된다. 또한, COVID-19와 같은 새로운 감염증에 대한 치료약의 개발 기간을 단축함으로써 모두가 체감할 수 있는 보건 및 경제적 가치를 창출할 수 있을 것으로 기대된다.
용어해설
De Novo Drug Design 한정된 화합물 라이브러리에서 신약 후 보물질을 스크리닝하는 전통적인 방식과 달리 인공지능 기반의 생성 모델과 특성 예측 모델을 이용해 원하는 물리화학적 및 약리 학적 특성을 갖는 후보물질을 새롭게 만들어 내는 기술
약어 정리
ADME
Absorption, Distribution, Metabolism, Excretion
DNN
Deep Neural Network
GAN
Generative Adversarial Network
QSAR
Quantitative Structure-Activity Relationship
RNN
Recurrent Neural Network
SMILES
Simplified Molecular Input Line Entry System
SOM
Self Organizing Map
SVM
Support Vector Machine
VAE
Variational Auto-Encoder
M. Steedman et al., "Intelligent biopharma: Forging the links across the value chain," Deloitte Insights, 2019, pp. 1-25, https://www2.deloitte.com/content/dam/Deloitte/ch/Documents/life-sciences-health-care/deloitte-ch-di-intelligent-biopharma.pdf
D. Weininger, "SMILES, a chemical language and information systems. 1. introduction to methodology and encoding rules," J. Chem. Inf. Comput. Sci., vol. 28, no. 1, 1988, pp. 31-36.
J. Arus-Pous et al., "Randomized SMILES strings improve the quality of molecular generative models," J. Cheminformatics, vol. 11, no. 71, 2019, pp. 1-13.
W. Jin et al., "Junction tree variational auto encoder for molecular graph generation," in Proc. 35th Int. Conf. Mach. Learn. (ICML), (Stockholm, Sweden), Jul. 2018.
M. Wang et al., "Deep learning approaches for de novo drug design: An overview," Current Opinion Struct. Biol., vol. 72, 2022, pp. 135-144.
C.A. Lipinski, "Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings," Adv. Drug Deliv. Rev., vol. 46, no. 1-3, 2001, pp. 3-26.
J. Mao et al., "Comprehensive strategies of machine learning based quantitative structure activity relationship models," iScience, vol. 24, no. 9, 2021, p. 103052.
M. Popova et al., "Deep reinforcement learning for de novo drug design," Sci. Adv., vol. 4, no. 7, 2018, eaap7885.
A. Zhavoronkov et al., "Deep learning enables rapid identification of potent DDR1 kinase inhibitors," Nat. Biotechnol., vol. 37, 2019, pp. 1038-1040.
J. Choi and J. Lee, "V-Dock: Fast generation of novel drug-like molecules using machine-learning-based docking score and molecular optimization," Int. J. Mol. Sci., vol. 22, no. 21, 2021, p. 11635.
M. Olivecrona et al., "Molecular de-novo design through deep reinforcement learning," J. Cheminform., vol. 9, no. 1, 2017, p. 48.
J. Guo et al., "DockStrem: A docking wrapper to enhance de novo molecular design," J. Cheminform., vol. 13, no. 1, 2021, p. 89.
J. Jumper et al., "Highly accurate protein structure prediction with AlphaFold," Nature, vol. 596, no. 7873, 2021, pp. 583-589.
P. Bryant et al., "Improved prediction of protein-protein interactions using AlphaFold2," Nat. Commun., vol. 13, no. 1, 2022, p. 1265.
V. Chenthamarakshan et al., "CogMol: Target-specific and selective drug design for COVID-19 using deep generative models," Proceedings of the 34th Conference on Neural Information Processing System(NeurIPS), 2020.
J. Born et al., "PaccMannRL: De novo generation of hitlike anticancer molecules from transcriptomic data via reinforcement learning," iScience, vol. 24, no. 4, 2021, p. 102269.
K. Lanevskij and R. Didziapetris, "Physicochemical QSAR analysis of passive permeability across Caco-2 monolayers," J. Pharm. Sci., vol. 108, no. 1, 2019, pp. 78-86.
H. Sun, "Highly predictive and interpretable models for PAMPA permeability," Bioorg. Med. Chem., vol. 25, no. 3, 2017, pp. 1266-1276.
F. Stefaniak, "Prediction of compounds activity in nuclear receptor signaling and stress pathway assays using machine learning algorithms and low-dimensional molecular descriptors," Front. Environ. Sci., vol. 3, 2015, p. 77.
E. Kim and H. Nam, "Prediction models for drug-induced hepatotoxicity by using weighted molecular fingerprints," BMC Bioinformatics, vol. 18, no. 227, 2017, pp. 25-34.
K. Ogura et al., "Support vector machine model for hERG inhibitory activities based on the integrated hERG database using descriptor selection by NSGA-II," Sci. Rep., vol. 9, no. 1, 2019, p. 12220.

- Sign Up
- 전자통신동향분석 이메일 전자저널 구독을 원하시는 경우 정확한 이메일 주소를 입력하시기 바랍니다.